Authors:
de Souza, Mariana L. [1] ; Rezende Junior, Celso de Oliveira [2] ; Ferreira, Rafaela S. [3] ; Espinoza Chavez, Rocio Marisol [2] ; Ferreira, Leonardo L. G. [1] ; Slafer, Brian W. [2] ; Magalhaes, Luma G. [1] ; Krogh, Renata [1] ; Oliva, Glaucius [1] ; Cruz, Fabio Cardoso [4] ; Dias, Luiz Carlos [2] ; Andricopulo, Adriano D. [1]
Abstract:
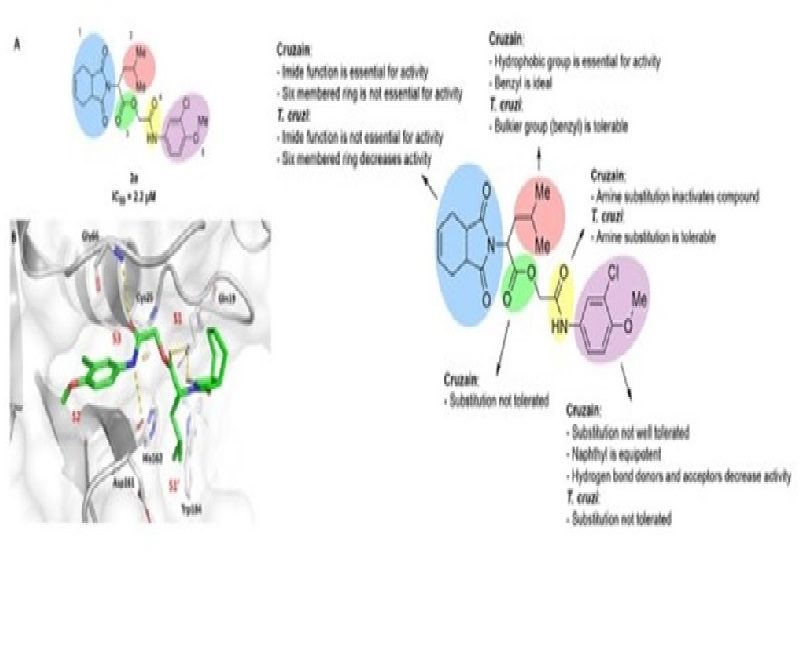
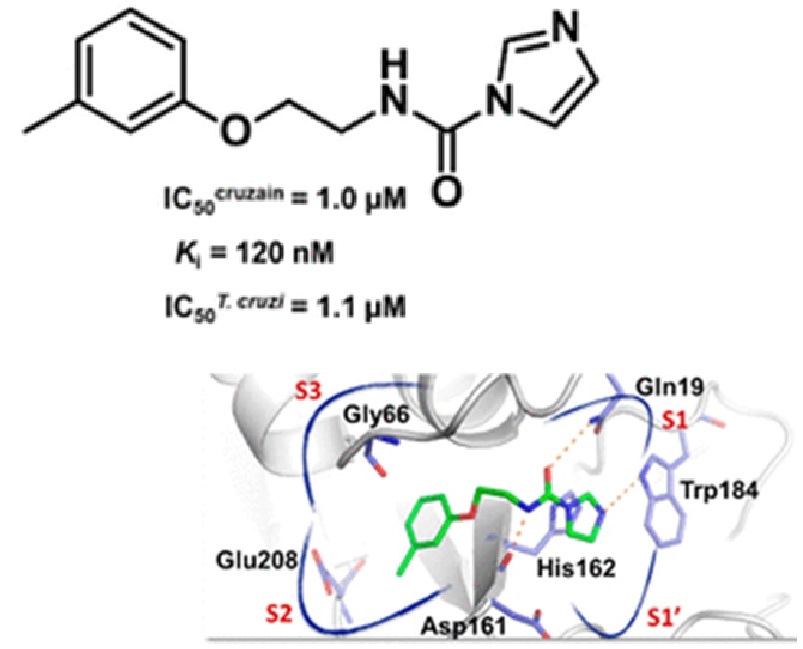
A virtual screening conducted with nearly 4 000 000 compounds from lead-like and fragment-like subsets enabled the identification of a small-molecule inhibitor (1) of the Trypanosoma cruzi cruzain enzyme, a validated drug target for Chagas disease. Subsequent comprehensive structure-based drug design and structure-activity relationship studies led to the discovery of carbamoyl imidazoles as potent, reversible, and competitive cruzain inhibitors. The most potent carbamoyl imidazole inhibitor (45) exhibited high affinity with a K-i value of 20 nM, presenting both in vitro and in vivo activity against T. cruzi. Furthermore, the most promising compounds reduced parasite burden in vivo and showed no toxicity at a dose of 100 mg/kg. These carbamoyl imidazoles are structurally attractive, nonpeptidic, and easy to prepare and synthetically modify. Finally, these results further advance our understanding of the noncovalent mode of inhibition of this pharmaceutically relevant enzyme, building strong foundations for drug discovery efforts.
1 Laboratory of Medicinal and Computational Chemistry, Physics Institute of Sao Carlos, University of Sao Paulo, Sao Carlos – SP, 13563-120, Brazil
2 Institute of Chemistry, State University of Campinas, Campinas – SP, 13084-971, Brazil
3 Department of Biochemistry and Immunology, Federal University of Minas Gerais, Belo Horizonte – MG, 31270-901, Brazil4Department of Pharmacology, Federal University of Sao Paulo, Sao Paulo – SP, 04023-062, Brazil
4 Departmente of Pharmacology, Federal University of São Paulo, BR-04023062 Sao Paulo, SP – Brazil
Link to article: https://pubs.acs.org/doi/10.1021/acs.jcim.9b00802